PRINCIPES TECHNIQUES DE LA CMF
Les principes de la cytométrie sont résumés dans la figure 1.
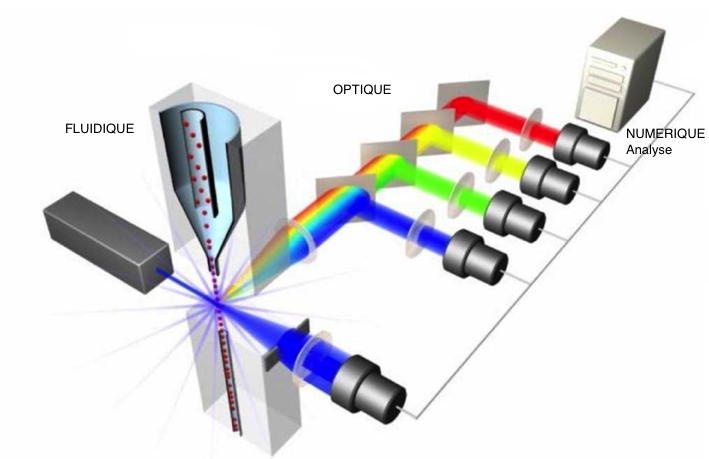
Figure 1 : Principe simplifié d'un cytomètre en flux
Pour fonctionner un cytométre en flux nécessite une combinaison
de:
Fluidique: Pour introduire et canaliser les cellules.
Optique: Une source d’excitation et de récupération des signaux,
Electronique: Pour convertir les signaux optiques en des signaux
électroniques proportionnels et les numériser pour les analyser
avec un ordinateur.
LES ECHANTILLONS CELLULAIRES
Les cellules doivent être mises
en suspension pour pouvoir être analysées. L’analyse du sang ne
pose aucun problème les cellules étant déjà en suspension. Par
contre les tissus cellulaires doivent être dissociés et les
agrégats éliminés afin de pouvoir être analysés.
LE CENTRAGE HYDRODYNAMIQUE
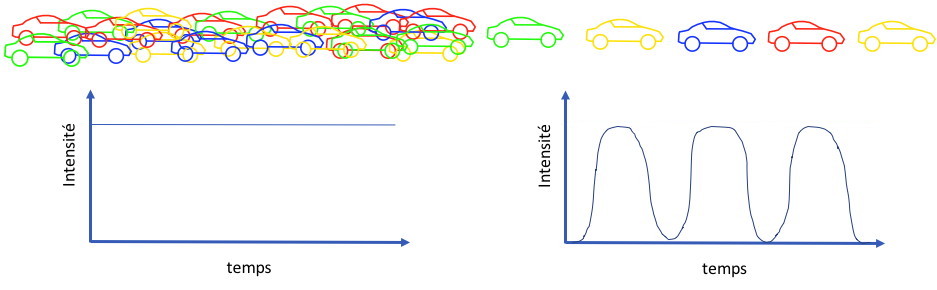
Imaginez devoir compter, sur une
autoroute avec de très nombreuses voies, les vehicules de
différentes couleurs (Figure 2). Si la circulation est chargée et
toutes les voies utilisées, il vous sera impossible d'effectuer
cette tâche. Pour y parvenir, vous devez canaliser les vehicules
sur une seule voie et, ainsi, il vous sera possible d'effectuer
une analyse précise du nombre de véhicules de chaque couleur.
Figure 2 : Principe de
comptage d'un cytomètre
Dans un cytomètre en flux il faut procéder de la même façon. Les
cellules sont amenées au centre de la buse de mesure et alignées
les unes derrière les autres (au moyen du sytème de centrage
hydrodynamique de l’échantillon) afin d’être excitées une par une
avec le faisceau lumineux. Le liquide de gaine subit une
accélération progressive ce qui entraîne un étirement du liquide
échantillon et ainsi aligne les cellules au centre du jet (Figure
3).
 Figure
3 : Principe du centrage hydrodynamique
Figure
3 : Principe du centrage hydrodynamique
Attention, lorsque l'échantillon de départ est faiblement
concentré, l'utilisateur d'un cytomètre a tendance à augmenter la
pression sur celui-ci afin de diminuer le temps d'acquisition.
Malheureusement, cela a pour effet de diminuer la précision du
centrage de l'échantillon et entraine une dispersion des mesures
puisque les cellules ne passeront pas nécessairement dans la zone
de focalisation de la source lumineuse (Figure 4). Cela est
facilement visualisé en passant des billes de calibration à
différentes pressions, leur pic s'élargit au fur et à mesure de
l'augmentation de pression (augmentation du coefficient de
variation). Cet effet est particulièrement préjudiciable aux
mesures de cycle cellulaire où la finesse des pics est un gage de
qualité de la mesure. Il est donc préférable d'augmenter la
concentration de l'échantillon plutôt que d'augmenter la pression
sur celui-ci. Bien évidemment, il faut aussi tenir compte des
limites de traitement des informations de l'appareil. De même en
augmentant trop la vitesse, on peut se retrouver dans le cas de
figure où plusieurs particules passent au même moment devant la
source lumineuse comprométant la fiabilité des mesures.

Figure 4 : Effet de
l'augmentation de pression sur l'échantillon sur la mesure du
signal
CIRCUITS OPTIQUES
La source d’excitation lumineuse
doit permettre une illumination des colorants à une longueur
d’onde proche de leur maximum d’absorption. Elle doit être
puissante, stable et nécessite une bonne focalisation.
Deux types de sources sont actuellement utilisées:
•Les lasers (les plus fréquemment utilisés) qui présentent un
grand nombre d’avantages: puissance, stabilité, finesse du
faisceau. Les lasers ont des spectres d’émission discontinus
(Figure 5).
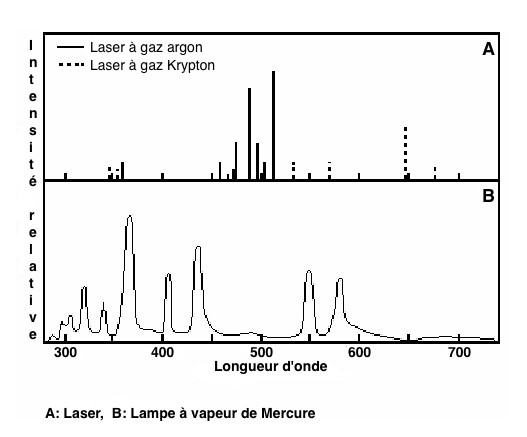
Figure 5 : principales sources lumineuses utilisées en
cytométrie en flux
Les lasers à ions d'argon sont maintenant remplacés par des diodes
laser nécessitant des contraintes d'installation moindres:


La présence de plusieurs lasers
de type différents permet de multiplier le nombre de fluorochromes
aux caractéristiques spectrales différentes.
Les lampes à vapeur de mercure ou au xénon étaient aussi
utilisées. La focalisation est moindre que dans le cas du laser,
mais le spectre est assez large et leur coût est limité.
Quand il y a plusieurs lasers au sein d'un cytomètre, ceux-ci
peuvent être disposés de 2 façons. Soit ils sont tous focalisés au
même endroit, on dit alors qu'ils sont colinéaires, soit ils sont
décalés dans l'espace (la cellule passe successivement devant
chaque laser) on dit alors qu'ils sont décalés.
L'intérêt des lasers décalés est qu'il permet, pour des
fluorochromes ayant des longueurs d'ondes d'excitation différentes
mais présentant des longueurs d'onde d'émission équivalentes
d'être mesurés indépendamment ce qui n'est pas possible avec des
lasers colinéaires (Figure 6). Les lasers colinéaires entrainent
donc des contraintes plus importantes dans le choix des
fluorochromes.
 Figure
6 : Configuration des lasers, décalés et colinéaires,
exemple du 7AAD et de l'APC
Figure
6 : Configuration des lasers, décalés et colinéaires,
exemple du 7AAD et de l'APC
COLLECTE DE LA LUMIERE EMISE
Les différents signaux optiques
émis par la cellule doivent être focalisés, séparés, puis
acheminés vers des systèmes de détection, photomultiplicateurs ou
photodiodes. Ils sont pour cela, sélectionnés par différents
circuits optiques, composés d’une alternance de miroirs et de
filtres.
Un miroir est une surface réfléchissante. Suivant le traitement de
sa surface, on peut obtenir trois types de miroirs: passe-haut,
passe-bas et passe bande (Figure 7, 8). De plus, les longueurs
d’onde réfléchies varient aussi avec l’angle formé par le rayon
incident et la surface du filtre.

Figure 7 : Les différents types de filtres utilisés en
cytométrie (R: Réflection, : longueur d'onde)
 Figure
8 : Les différents types de filtres utilisés en cytométrie
Figure
8 : Les différents types de filtres utilisés en cytométrie
Si les longueurs d'onde non
réfléchies sont transmises, on obtient ainsi des miroirs
dichroïques.
Pour les filtres, on retrouve en transmission les mêmes courbes
que pour les miroirs, par contre les longueurs d'onde non
transmises ne sont pas réfléchies. Elles sont soit absorbées soit
détruites.
Après avoir traversé cette succession de miroirs et de filtres, la
lumière est recueillie et transformée en signal électrique
directement proportionnel à la lumière reçue par un
photomultiplicateur ou une photodiode (Figure 9 et 10).
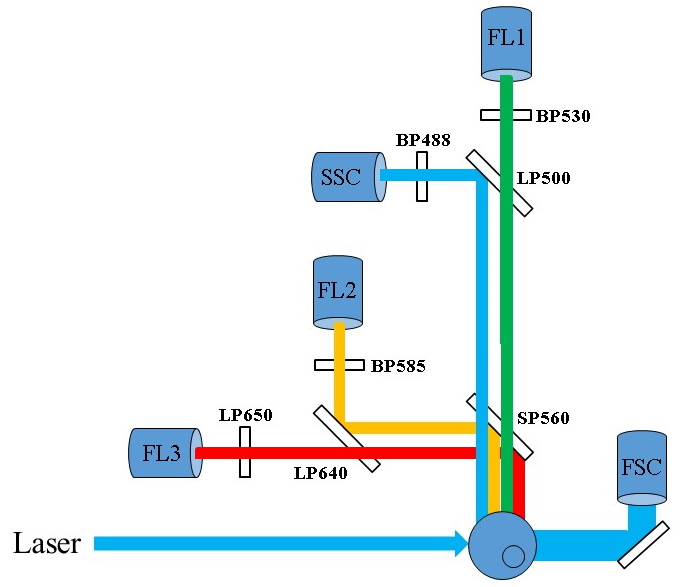
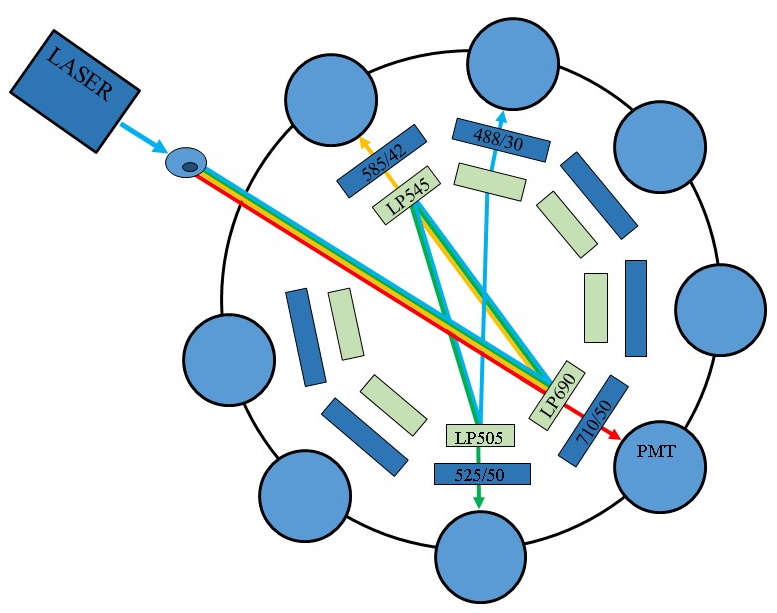 Figure
9 : Exemple de trajet optique dans un cytomètre en flux (©Becton
Dickinson)
Figure
9 : Exemple de trajet optique dans un cytomètre en flux (©Becton
Dickinson)
Flow Cytometry Experiment Planning Steps
Figure
10 : Exemple de trajet optique dans un cytomètre en flux
spectral (©Cytek)
Flow Cytometry Experiment Planning Steps
Flow Cytometry Experiment Planning Steps
LES SIGNAUX RECUEILLIS
Les signaux optiques recueillis ont une intensité corrélée avec des
propriétés cellulaires (tableau 1).
| PARAMETRE |
SIGNIFICATION |
UTILISATION |
| Diffusion de la lumière aux petits
angles |
Proportionnel au diamètre cellulaire |
Identification morphologique des
cellules |
| Diffusion de la lumière à angle droit |
Proportionnel au contenu cellulaire |
Identification morphologique des
cellules |
| Fluorescences |
Proportionnelles
à l’intensité de marquage |
Marqueurs
cellulaires, ADN, ARN, fonctions cellulaires... |
Tableau 1:
Signification des principaux signaux obtenus en cytométrie en
flux.
La lumière diffusée
La lumière diffusée renseigne sur
la morphologie et la structure de la cellule:
Si la diffusion de la lumière est mesurée dans l’axe du rayon
incident, l’intensité du signal peut être corrélée avec la taille
et la viabilité cellulaire.
Sous un angle de 90°, la mesure correspond à la structure
intracellulaire de la cellule (réfringence du cytoplasme,
morphologie, rapport nucléo-cytoplasmique).
L’utilisation simultanée de ces deux paramètres permet de
distinguer, dans un sang périphérique par exemple, les plaquettes,
les lymphocytes, les monocytes et les polynucléaires (Figure 11).

Figure 11 : Utilisation de la double diffusion de la lumière
pour distinguer les diverses sous populations sanguines (FSC:
diffusion aux petits angles, SSC diffusion aux grands angles).
Le seuil
Le choix du seuil ou threshold
permet d'éliminer de l'analyse les particules de valeurs
inférieures au seuil choisi sur le paramètre choisi. Par exemple,
un seuil sur la taille permettra d'éliminer les particules les
plus petites et ainsi d'éviter de polluer le fichier d'acquisition
avec celles-ci. Cela peut servir, par exemple, à éliminer les
plaquettes ou globules rouges d'une acquisition sur des
lymphocytes. Attention de ne pas choisir un seuil trop important
sous peine de risquer de ne plus voir certaines populations
cellulaires de taille intermédiaire ou même non fluorescentes si
l'on choisit de mettre un seuil sur une fluorescence. Le seuil sur
une fluorescence est utile pour analyser un cycle cellulaire
puisqu'il permet d'éviter de mesurer des cellules non
fluorescentes ou des petits fragments d'ADN.

La fluorescence émise
Cette fluorescence peut être
spontanée, mais le plus souvent, elle est apportée à la cellule
par un fluorochrome. Le fluorochrome absorbe l’énergie du LASER et
réemet l’énergie absorbée par vibration et dissipation de chaleur,
émission de photons d’une longueur d’onde plus élevée.
Un fluorochrome est caractérisé par son spectre lumineux
d'absorption et d'émission. La lumière réémise est toujours d'une
longueur d'onde supérieure à la longueur d'onde absorbée.
Les caractéristiques de quelques fluorochromes utilisés en CMF
sont résumées dans le Tableau 2 :
FLUOROCHROME
|
PROPRIETE
|
Excitation
Max*
|
Emission
Max@
|
Hoechst 33342
|
ADN
(A-T)
|
365
|
402
|
DAPI
|
ADN
(A-T)
|
357
|
451
|
Chromomycine A3
|
ADN
(G-C)
|
450
|
470
|
Iodure de Propidium
|
ADN
(intercalant)
|
536
|
620
|
Acridine Orange
|
ARN/ADN
|
492
|
527
db/630 sbrin
|
Alexa 488
|
Conjugaison
|
498
|
520
|
FITC
|
Conjugaison |
490
|
543
|
PE
|
Conjugaison |
490/565
|
578
|
PerCP
|
Conjugaison |
488
|
675
|
APC
|
Conjugaison |
642
|
660
|
PE-Cy5.5
|
Conjugaison |
490/565 |
693
|
PE-Cy7
|
Conjugaison |
490/565 |
778
|
Brilliant Violet 510
|
Conjugaison |
405
|
510
|
| Brilliant Violet 650 |
Conjugaison |
405
|
650
|
| GFP |
Gene
Reporter |
488 |
510 |
| mCherry |
Gene
Reporter |
585
|
610
|
dTomato
|
Gene
Reporter |
554
|
580 |
Rhodamine 123
|
potentiel
mitochondrial
|
505
|
534
|
BCECF-AM
|
pH
|
440
|
530
|
SNARF-1
|
pH/traceur
|
488
|
580/630
|
CFSE
|
traceur
|
498
|
518
|
Cascade blue
|
traceur
|
399
|
423
|
Fura Red
|
Ca2+
|
450-500
|
660
|
Indo1-AM
|
Ca2+ |
331
|
405/480
|
Fluo-3
|
Ca2+ |
506
|
526
|
Tableau
2 : Caractéristiques de quelques fluorochromes utilisés en
cytométrie. (*: longueur d'onde d'excitation, @: longueur d'onde
d'émission).
Il existe
- des Fluorochromes à affinité propre pour un constituant
cellulaire: par exemple pour les mesures d’ADN, d’ARN, de
protéines, du pH, de calcium contenus dans la cellule.
- des Fluorochromes couplés à un ligand spécifique. Cette
spécificité peut être amenée par un couplage du fluorochrome à un
anticorps ou un ligand spécifique d’un constituant cellulaire.
Attention car les fluorochromes n'ont pas tous le même rendement
de fluorescence et n'auront donc pas les mêmes capacités de
détection. Pour le démontrer, il 'suffit' de coupler le même
anticorps avec différents fluorochromes et de comparer l'intensité
des marquages sur les mêmes cellules (
Figure 12).

Figure 12 : Etude de la sensibilité des fluorochromes par un
marquage avec un anticorps CD8 (©Beckman
Coulter : O Jaen) CONVERSION DES SIGNAUX
Les signaux optiques sont
convertis en signaux électriques par les photomultiplicateurs. Ces
signaux électriques ont une valeur comprise le plus souvent entre
0 et 10 volts (signal analogique). L’ordinateur ne peut traiter
ces données que si elles sont sous forme binaire (signal digital).
Le rôle du convertisseur analogique-digital est de convertir,
comme son nom l’indique, un signal analogique (valeur continue) en
signal digital (valeur discontinue) assimilable par l’ordinateur.
C’est à dire de transformer une valeur comprise entre 0 et 10
volts en une valeur binaire comprise entre 0 et 1023 pour un
convertisseur 10 bits (210) (Figure 13). Les
nouvelles machines convertissent maintenant les signaux sur
plusieurs millions de canaux.

Figure 13: Principe de fonctionnement d'un convertisseur
PRESENTATION DES RESULTATS
Les valeurs numériques issues des
convertisseurs sont stockées par l’informatique et présentées sur
les écrans des cytomètres sous deux formes :
•des histogrammes monoparamétriques où l’axe des abscisses
représente l’intensité du signal analysé et l’axe des ordonnées le
nombre de cellules, les axes peuvent être linéaires ou
logarithmiques (
Figure 14).
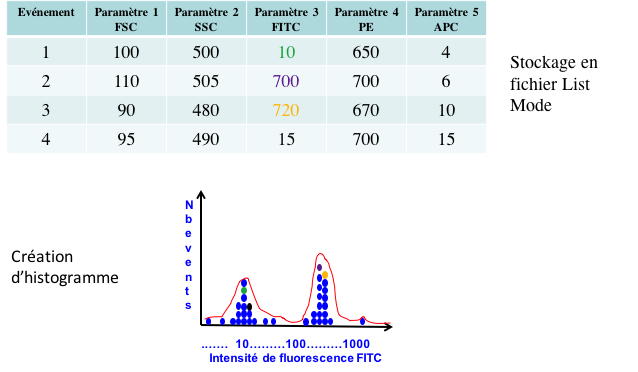
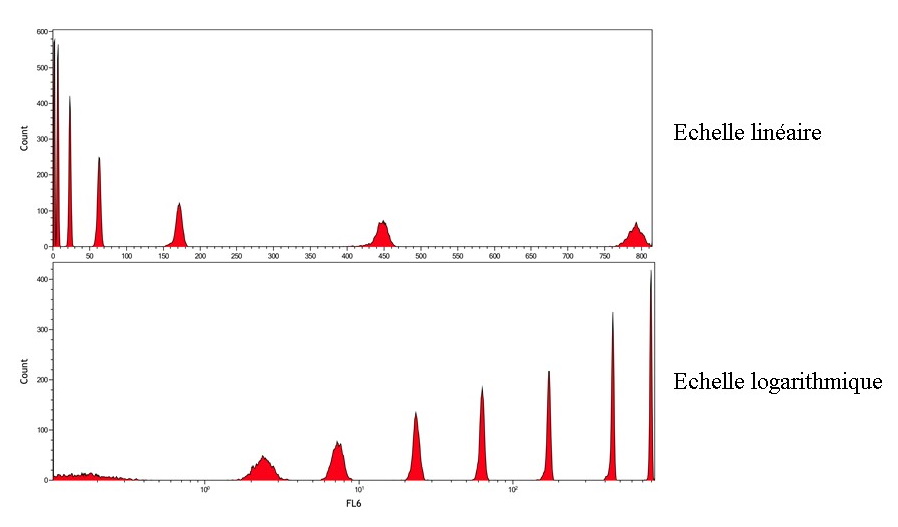
Figure 14 : Obtention d'un histogramme monoparamétrique.
•des histogrammes biparamétriques ou cytogrammes présentant deux
signaux simultanément (
Figure 15).

Figure 15 : Présentation des résultats en cytométrie en flux.